- Accueil
- Protection individuelle - Vêtements Chaussures
- Protection individuelle
EPI (Protection Individuelle), fournisseurs et fabricants Français
La protection individuelle (EPI) du personnel : un enjeu majeur
Règlement UE2016/425
L'EPI 'Equipement de Protection Individuelle est un dispositif ou un moyen destiné à être porté ou tenu par une personne en vue de la protéger contre un ou plusieurs risques susceptibles de menacer sa santé ou sa sécurité.
Le règlement européen 2016/425 traite des obligations des fabricants d’Equipements de Protection Individuelle.
Exigences générales CE
- Ergonomie, Innocuité des EPI (matériaux, santé,...), Facteur de confort et d’efficacité (adaptation à la morphologie, légèreté, solidité,...), Notice technique des EPI
Exigences communes CE
- Réglages, vieillissement, intervention rapide...
Exigences spécifiques à des risques particuliers CE
- Glissade, chutes de hauteur, chaleur, froid, rayonnement...
Les organismes notifiés : indépendants et notifiés par l’Etat.
Ils valident les produits testés selon les normes, si elles existent, pour les normes européennes.
Ils vérifient que l’EPI passe les tests de performances exigés, mais vérifient également le marquage du produit et des emballages selon les exigences, le contenu de la notice technique du fabricant, la mise en place d’un système de qualité pour contrôler les EPI de catégorie 3.
Plusieurs fabricants peuvent avoir le même numéro d’organisme notifié. Chaque organisme notifié à son propre numéro et peut auditer plusieurs fabricants.
Que dis le règlement UE ?
Le Règlement UE 2016/425 du 9 mars 2016 définit les catégories d’EPI dans son annexe 1 :
EPI catégorie 1 Risques mineurs
EPI catégorie 2 Risques intermédiaires
EPI catégorie 3 Risques mortels, dommages irréversibles
Choisissez parmi les protection individuelle
Les produits indispensables
Aucun produit ne correspond à votre recherche


-100%
Ref: 40.632
1,25€ HT
1,25€ HT
Blouse jetable de qualité supérieure en polypropylène 40 grammes par m2. Blouse d’hygiène avec fermeture …
3241 En stock
 Supprimer des favoris
Supprimer des favoris


-100%
Ref: 888255
0,85€ HT
0,85€ HT
Blouse jetable en polypropylène blanc de 30 gr/m2.
Cousue élastiquée au poignet et fermée par 4 boutons …
2305 En stock
Supprimer des favoris

-100%
Ref: 888249
2,40€ HT
2,40€ HT
Charlotte à clips en non tissée de polypropylène blanche . Cette charlotte dispose d’un élastique thermoc…
994 En stock
Supprimer des favoris

-100%
Ref: 888832
2,60€ HT
2,60€ HT
Combinaison en SMS polypropylène non tissé 3 couches: Spunbonded Meltblown Spunbonded.
Capuche en 3 pièc…
840 En stock
Supprimer des favoris

-100%
Ref: WL-CB
2,45€ HT
2,45€ HT
Combinaison jetable coloris bleu catégorie III type 5 et 6 offrant un confort maximum (haute perméabilité…
809 En stock
Supprimer des favoris

-100%
Ref: WL-PMP
4,40€ HT
4,40€ HT
Les coutures recouvertes assurent une éctanchéité contre les particules dilués, et les pesticides dilués,…
732 En stock
Supprimer des favoris

-100%
Ref: WL-C1
3,65€ HT
3,65€ HT
Weecover max 1 de Weesafe est une combinaison jetable spéciale amiante à coutures thermocollées à plat po…
718 En stock
Supprimer des favoris

-100%
Ref: WL-PMG
5,35€ HT
5,35€ HT
Permet d’assurer la protection des opérateurs pendant les opérations de traitements et met en sécurité le…
634 En stock
Supprimer des favoris

-100%
Ref: WL-B
3,80€ HT
3,80€ HT
Combinaison jetable avec dos aéré Weeback catégorie III type 5 et 6. Le dos aéré de la combinaison laisse…
622 En stock
Supprimer des favoris

-100%
Ref: 70.101
2,65€ HT
2,65€ HT
Modèle clips blanche adaptée à l’usage en collectivité, restauration collective, industrie, laboratoire, …
434 En stock
Supprimer des favoris

-100%
Ref: WL-J1
7,85€ HT
7,85€ HT
Modèle offre une double protection avec une première fermeture à zip recouverte avec un rabat, et une deu…
193 En stock
Supprimer des favoris

-100%
Ref: WA-KAN
30,90€ HT
30,90€ HT
Kit de protection jetable pour travaux en présence d’amiante. Ce kit est totalement adapté aux travaux de…
106 En stock
Supprimer des favoris

-100%
Ref: 40.507
2,90€ HT
2,90€ HT
Blouse jetable microporeuse SMS antistatique PP+PE avec col. Cette blouse est un équipement de protection…
102 En stock
Supprimer des favoris

-100%
Ref: 40.453EU
4,95€ HT
4,95€ HT
Bouse visiteur jetable pour isolation et limiter les contaminations croisées.
Version renforcée avec gra…
98 En stock
Supprimer des favoris

-100%
Ref: 888118
68,15€ HT
68,15€ HT
Notre combinaison jetable blanche et en polypropylène grammage 40grs pour travaux professionnels tels qu…
75 En stock
Supprimer des favoris

-100%
Ref: J40451
7,30€ HT
7,30€ HT
Blouse à usage unique jetable d’isolation en polypropylène coloris verte. Blouse portée par le personnel …
69 En stock
Supprimer des favoris
-100%
Ref: 70.103
3,40€ HT
3,40€ HT
A clips verte adaptée à l’usage en collectivité, restauration collective, industrie, laboratoire, industr…
66 En stock
Supprimer des favoris

-100%
Ref: 0510807
4,55€ HT
4,55€ HT
Charlotte ronde usage unique coloris blanche. Charlotte en polypropylène non tissé avec élastique cousu s…
64 En stock
Supprimer des favoris
-100%
Ref: 70.102
3,40€ HT
3,40€ HT
Charlotte jetable clips bleue adaptée à l’usage en collectivité, restauration collective, industrie, labo…
51 En stock
Supprimer des favoris

-100%
Ref: 70.001
5,05€ HT
5,05€ HT
Ronde et de coloris blanche est en polypropylène avec une confection cousue et élastiquée.Elle est facile…
45 En stock
Supprimer des favoris

-100%
Ref: J40452
7,25€ HT
7,25€ HT
Blouse à usage unique jetable d’isolation en polypropylène coloris blanche. Blouse portée par le personne…
44 En stock
Supprimer des favoris
-100%
Ref: 888260
8,35€ HT
8,35€ HT
Charlotte jetable en polypropylène blanc avec visière.
Adaptée à la méthode HACCP.
Charlotte casquette …
40 En stock
Supprimer des favoris

-100%
Ref: C12001
9,25€ HT
9,25€ HT
Modèle clip coloris blanche fabriquée en France. Fabriquée en non-tissé spunbonded 100% polypropylène.
C…
38 En stock
Supprimer des favoris

-100%
Ref: 0614828BLR
35,40€ HT
35,40€ HT
Blouse visiteur bleue en polypropylène non tissé avec fermeture par lanières dos et cou. Conditionné en s…
35 En stock
Supprimer des favoris

-100%
Ref: 05108081
4,75€ HT
4,75€ HT
Dispose d’un élastique thermocollé sans latex pour éviter toute allergie. Modèle à clips en non tissée de…
29 En stock
Supprimer des favoris

-100%
Ref: 88810500
37,30€ HT
37,30€ HT
Kit de protection jetable pour travaux en présence d’amiante. Combinaison non tissé SMS blanche.
Le kit …
20 En stock
Supprimer des favoris

-100%
Ref: 888251
6,20€ HT
6,20€ HT
Protéger la têtes avec un calot en papier jetble permet de réduire le risque de chute de cheveux dans les…
19 En stock
Supprimer des favoris
-100%
Ref: 70.112
10,70€ HT
10,70€ HT
Modèle avec double élastique « spécial confort » qui assure à l’utilisateur un excellent maintien. Systèm…
12 En stock
Supprimer des favoris

-100%
Ref: C13600
30,15€ HT
30,15€ HT
Bonnet universel jetable chirurgien en viscose pour domaine hospitalier et médical. Ce bonnet jetable est…
11 En stock
Supprimer des favoris

-100%
Ref: J40453
7,25€ HT
7,25€ HT
Blouse à usage unique jetable d’isolation en polypropylène coloris bleu ciel. Blouse portée par le person…
8 En stock
Supprimer des favoris

-100%
Ref: 888274
4,80€ HT
4,80€ HT
Toque de cuisinier jetable papier crépé extra blanc. Fond ajouré qui améliore la respirabilité de la toqu…
8 En stock
Supprimer des favoris

-100%
Ref: 888272
68,80€ HT
68,80€ HT
Kit visiteur comprenant une blouse en polyéthylène avec capuche et boutons pressions, une charlotte, un m…
7 En stock
Supprimer des favoris

-100%
Ref: C12101
9,10€ HT
9,10€ HT
Utilisable dans les environnements pharmaceutique, industriel, agro-alimentaire, médical...L’assemblage e…
7 En stock
Supprimer des favoris

-100%
Ref: C12201
9,10€ HT
9,10€ HT
Utilisable dans les environnements pharmaceutique, industriel, agro-alimentaire, médical...Sa grande exte…
5 En stock
Supprimer des favoris

-100%
Ref: C12103
10,95€ HT
10,95€ HT
Modèle clip coloris bleue fabriquée en France. Avec élastique synthétique sans latex sans ourlet pour un …
4 En stock
Supprimer des favoris

-100%
Ref: C13700
28,30€ HT
28,30€ HT
Bonnet universel jetable type chirurgien en viscose pour domaine hospitalier et médical. Ce bonnet jetabl…
4 En stock
Supprimer des favoris

-100%
Ref: 888273
132,30€ HT
132,30€ HT
Blouse visiteur polyéthylène avec capuche épaisseur 15 microns.
Longueur = 160 mm (avec capuche) / Large…
4 En stock
Supprimer des favoris

-100%
Ref: 20.909S
49,90€ HT
49,90€ HT
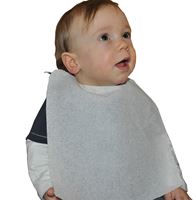
Bavoir jetable bébé en ouate blanc / PE 24 gr/m2 petit modèle fabrication Française. e bavoir enfant Safe…
3 En stock
Supprimer des favoris
-100%
Ref: 20.910S
98,60€ HT
98,60€ HT
Bavoir jetable bébé en ouate blanc et polyéthylène grand modèle fabriqué en France par Kolmi Medicom.Dime…
3 En stock
Supprimer des favoris


-34%
Ref: 178697
129,00€ HT
195,45€ HT
Bavoir Dunicel sur le thème de la mer.
Ce bavoir est idéal pour les restaurants à thématique fruits de l…
2 En stock
Supprimer des favoris

-100%
Ref: A69955
175,35€ HT
175,35€ HT
Blouse d’isolation imperméable pour protéger les professionnels de la santé et les patients d’une infecti…
2 En stock
Supprimer des favoris

-47%
Ref: 20927
94,10€ HT
176,40€ HT
Bavoir jetable bleu ouate / PE 35 gr/m2 .
Dimensions: 49x38 cm.
Très pratique, le bavoir est résistant…
1 En stock
Supprimer des favoris
-100%
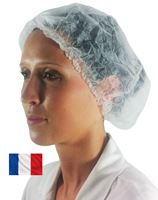
Ref: C12004
11,20€ HT
11,20€ HT
Elastique synthétique sans latex sans ourlet pour un meilleur confort des agents. Modèle clip coloris bla…
60 Dispo le 31/12
Supprimer des favoris
Focus sur protection individuelle
EPI ce qu'il faut savoir
Règlement UE 2017/745 équipement protection individuelle
La Directive Européenne 93/42 relative aux dispositifs médicaux a laissé place depuis le mois de Mai 2021 au Règlement (UE) 2017/745
commun pour l’ensemble des états membres.
• Les raisons d'un changement
La Directive Européenne 93/42/CEE est un texte réglementaire rédigé par l’Union Européenne, appliqué depuis 1993, donnant des indications communes concernant la mise sur le marché des dispositifs médicaux. Cette directive, applicable à l’ensemble des états membres de l’Union Européenne, a été enrichie par d’autres directives depuis près de 25 ans. Chacune de ces directives a été transposée dans la législation de chaque Etat membre de l’Union Européenne. Cela laisse ainsi libre cours à l’interprétation de chaque Etat membre, concernant les obligations de mise sur le marché des dispositifs médicaux.
Le parlement Européen a voté en Avril 2017 la mise en place d’un nouveau texte visant à unifier l’ensemble des acteurs du dispositif médical sous un seul et même règlement, plus complet suivant le contexte technologique actuel.
Ce nouveau règlement a pour but d’améliorer la traçabilité et la transparence au niveau Européen, mais aussi de pouvoir surveiller de façon accrue les organismes notifiés. Le règlement sur les dispositifs médicaux est entré en vigueur le 26 mai 2017, et est appliqué depuis le 26 mai 2021.
Cette transition doit être appliquée par les entreprises mais aussi par les organismes notifiés.
Quels changements pour les EPI ?
Le règlement modifie le champ des dispositifs médicaux, renforce considérablement leurs critères et procédures d’évaluation, ainsi que les responsabilités des différents acteurs.
? des démarches renforcées : le règlement n'est plus transposé dans le code de santé de chaque pays de l’Union Européenne mais devra être appliqué en l’état. Cela permet d’éviter les disparités entre les différents organismes notifiés et Etats membres au niveau des exigences à respecter.
? amélioration du suivi - EUDAMED : Parmi les changements notables, un nouveau système de surveillance post-certification est mis en application dès le 26 Mai 2021 afin d’identifier chaque dispositif médical suivant son propre code appelé «UDI» (Unique Device Identification). L’ensemble de ces données est répertorié au sein de la nouvelle base de données européenne appelée EUDAMED.
Ce «numéro de série» propre à chaque dispositif médical permettra une traçabilité très précise.
Cette nouveauté sera très utile lors des rappels de certains dispositifs et notamment dans le but d’évaluer le plus rapidement possible le nombre de personnes concernées par l’incident.
? évaluations cliniques : Un cadre plus précis en matière d’évaluations et d’investigations cliniques pour les DM avant la mise sur le marché.
Protection individuelle pour les dispositifs médicaux
Les DM sont répartis en quatre classes en fonction de leur niveau de risque :
- Classe I Faible degré de risque
- Classe IIa Degré moyen de risque
- Classe IIb Potentiel élévé de risque
- Classe III Potentiel très sérieux de risque
Les règles à suivre pour déterminer la classe du DM sont énoncées dans l’Annexe VIIII du règlement 2017/745 et prennent en compte la durée d’utilisation, le caractère invasif ou non et le type d’invasivité, la possibilité ou non de réutilisation, la visée thérapeutique ou diagnostique et la partie du corps concernée. Les DM de classe I non stériles ou n’ayant pas de fonction de mesurage sont autocertifiés par le fabricant. Pour les autres dispositifs; l’intervention d’un organisme notifié est systématique pour le processus de certification CE.